







Research
|
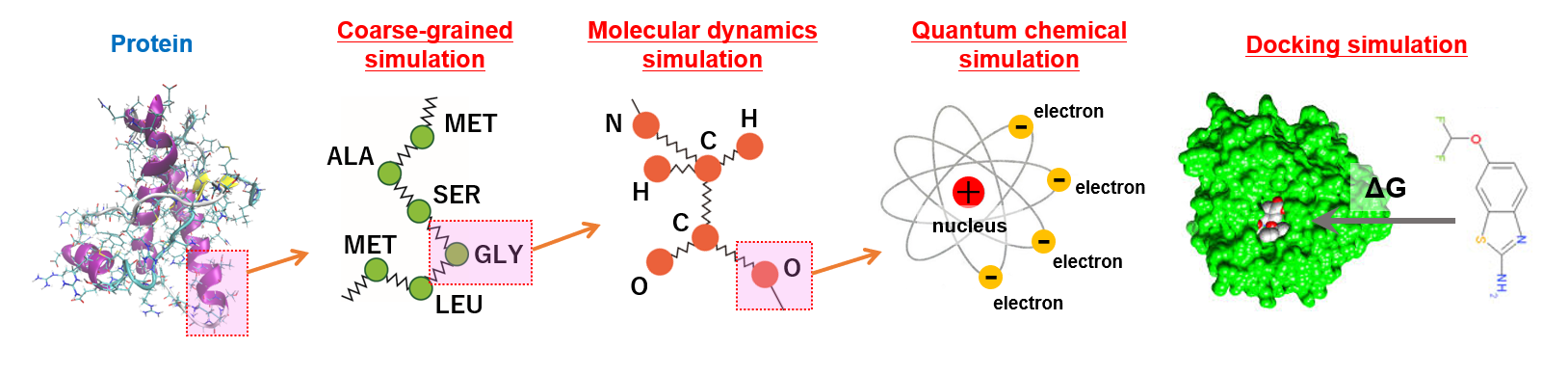
Computational chemistry is a research field in which various chemical phenomena are understood and predicted by solving the physical equations that molecules, atoms, or electrons obey. Generally, these physical equations are very complicated, so we use computers to carry out our research. In our laboratory, we hope to contribute to society by researching and solving various problems in the life sciences based on computational chemistry.
* Click on an item below to see its description, and click on an image to enlarge it. |
{kind=link}
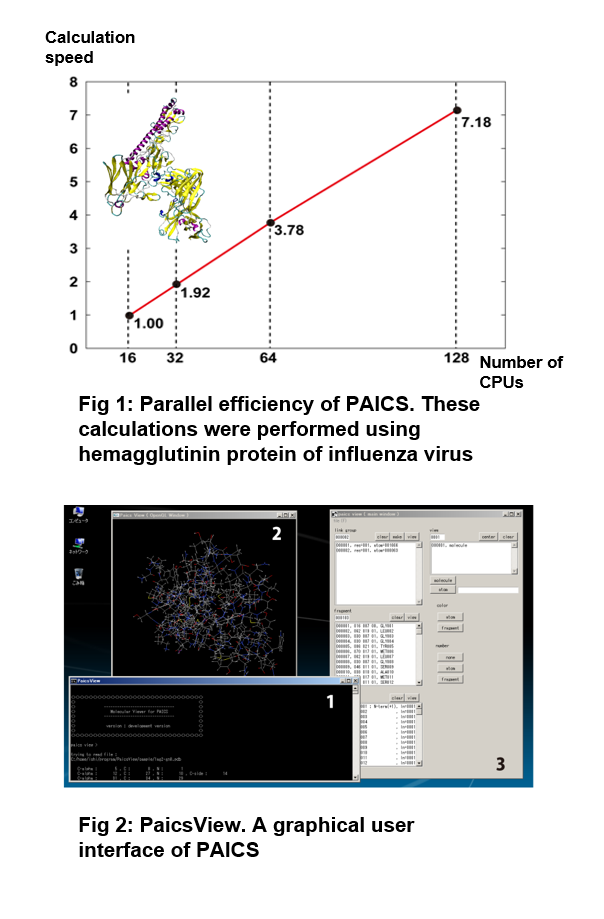
| In our laboratory, an original quantum chemical program “Parallelized Ab Initio Calculation System Based on FMO (PAICS)” is developed (http://www.paics.net/).This program is available to use the fragment molecular orbital (FMO) method, which enables us to perform quantum chemical calculations of large molecules such as biomolecules. In the FMO method, a target molecule is divided into small fragments, and only calculations of monomers and dimers for each fragment are required [1]. As a result, the computational efforts can be greatly reduced compared to the traditional quantum chemical methods. In addition, these monomer and dimer calculations are completely independent of each other, resulting in high parallel efficiency. In fact, PAICS is parallelized using Message Passing Interface (MPI) and is available to perform parallel computations using hundreds or thousands of CPUs. Fig. 1 shows the results of a benchmark on parallel efficiency. When the number of used CPUs increases by a factor of 8 (from 16 to 128), the computation speed increases by a factor of 7.18, indicating that high parallel efficiency is achieved. PaicsView, which is a user interface of PAICS, is also developed (Fig. 2). |
|
{kind=link}
|
References:
[1] K. Kitaura, et al, Chem. Phys. Lett., 313 (1999) 701. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
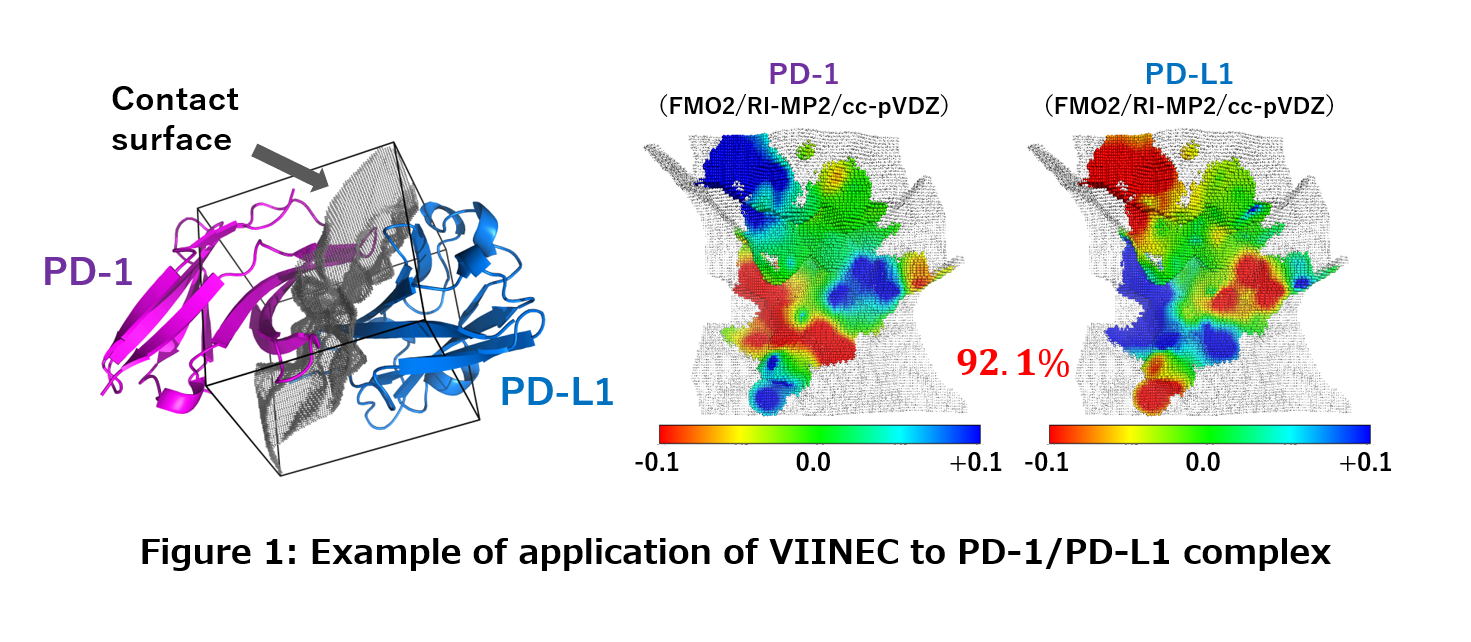
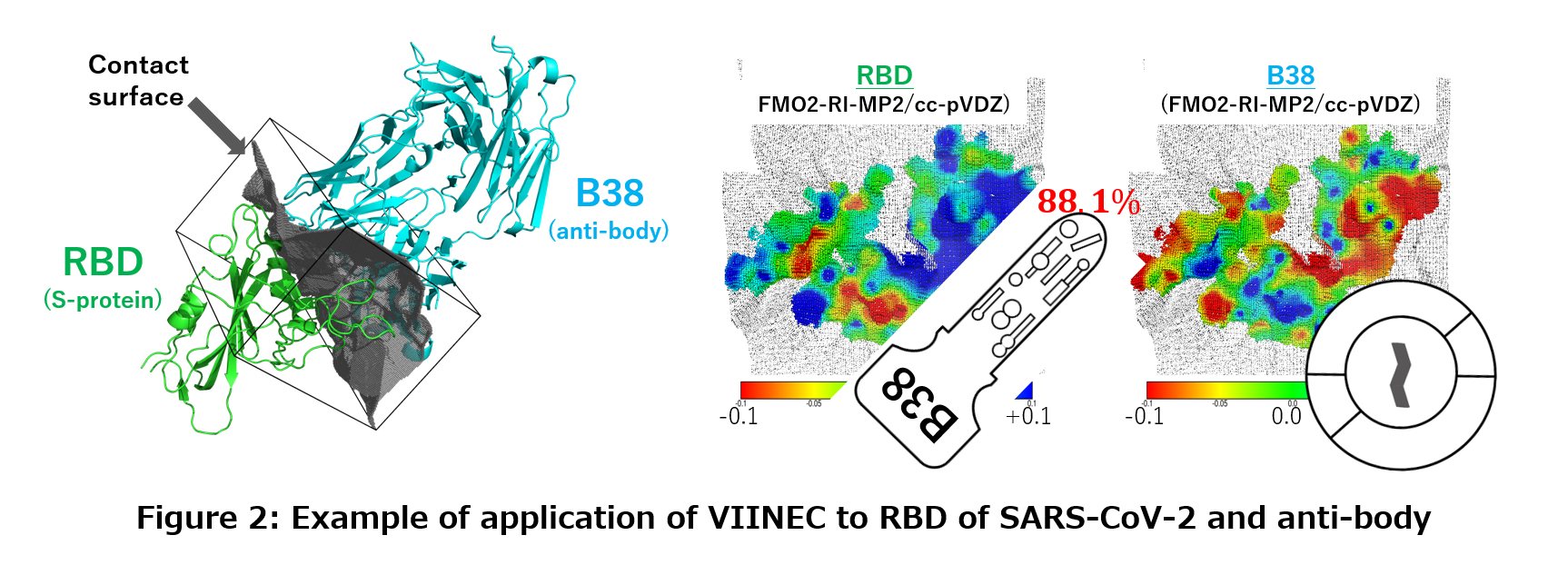
| As mentioned above, drug discovery for infectious diseases is a very important research issue, but even if new compounds are developed, in many cases pathogenic microorganisms that have acquired resistance emerge. In particular, RNA viruses, such as influenza, mutate very rapidly, and existing drugs may soon become useless. Therefore, we are also working on predicting resistance mutations to specific drugs using computers. |
|
Chemical reaction simulations involving the bond formation and annihilation require quantum chemical calculations that directly solve the equations of motion of electrons rather than classical calculations that use empirical potential functions. However, it is generally difficult to perform such calculations in aggregated systems such as solvents because of the huge computational time required. Therefore, we have developed a simulation method using the fragment molecular orbital method that drastically reduces the calculation time [1]. As a first application study, we performed a simulation of the hydrolysis reaction of methyl diazonium ion in an aqueous solution. In this case, all of the more than 150 water molecules and reacting molecules were treated quantum chemically, and the chemical reaction was successfully reproduced in the computer (see the movie on the right). |
|
参考文献:
[1] M. Sato, et al, J. Am. Chem. Soc. Comm. 130 (2008) 2396 |
|
In our laboratory, we address various problems in chemistry and life science by using computer simulations instead of conducting experiments. The computing resources currently managed by the laboratory are as follows (as of January 2026).
|
© 2018 Kagoshima University, Graduate School of Science and Engineering, laboratory of computational chemistry for life science