







����
|
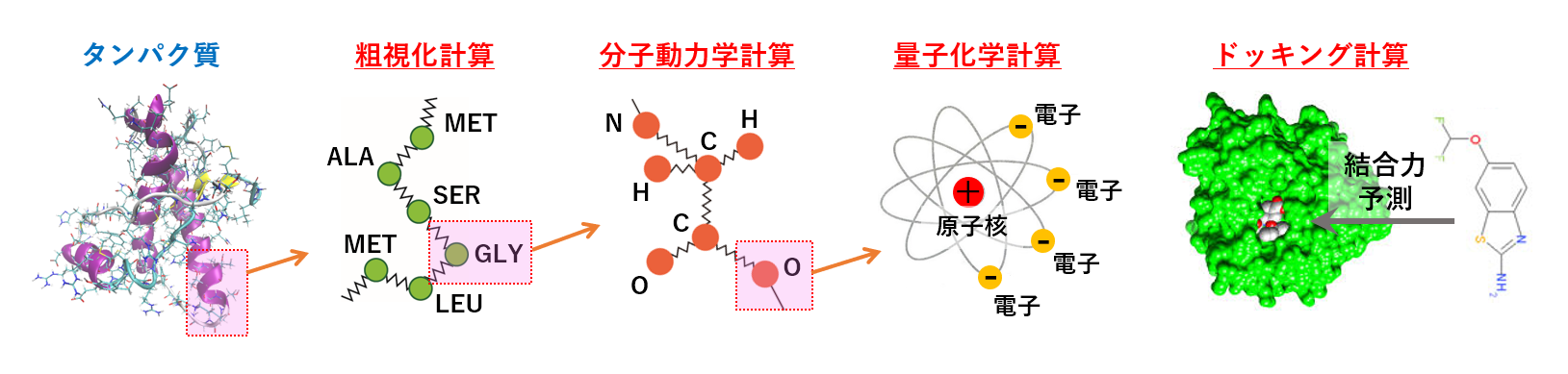
���q�⌴�q�������͓d�q���]���������������������ƂŁA�l�X�ȉ��w�I���ۂ𗝉��E�\�����錤��������u�v�Z���w�v�ƌĂт܂��B
�����̏ꍇ�A�����̕����������͔��ɕ��G�Ȃ̂ŁA�R���s���[�^�[�𗘗p���Č�����i�߂邱�ƂɂȂ�܂��B
��X�́u���� �v�Z���w �������v�ł́A�����Ȋw�Ɋւ��鏔�����v�Z���w�Ɋ�Â��Č����E�������邱�ƂŁA�Љ�ւ̍v�����ʂ����Ă��������ƍl���Ă��܂��B
�� �ȉ��̍��ڂ��N���b�N����ƁA�������\������܂��B�܂��A�摜���N���b�N����Ɗg��\������܂��B |
{kind=link}
|
��X�̌������ł́A�Ǝ��̗ʎq���w�v�Z�v���O�����uPAICS�v�̊J����i�߂Ă��܂�
(PAICS��HP)�B
PAICS�Ƃ������O�́uParallelized Ab Initio Calculation System Based on FMO�v�̓������������Ă��܂��B
���̃v���O�����ł́A���̕��q�̂悤�ȋ���ȕ��q�̗ʎq���w�v�Z���\�ɂ��邽�߂ɁA�u�t���O�����g���q�O���iFMO�j�@�v�ƌĂ��v�Z���@���̗p���Ă��܂��B
FMO�@�ł́A�v�Z�Ώۂ̕��q�������ȃt���O�����g�ɕ������A�t���O�����g�̒P�́i���m�}�[�j����уy�A�i�_�C�}�[�j�̌v�Z�݂̂���S�̂̕����ʂ��Z�o���܂�[1]�B
����ɂ��A�ʏ�̗ʎq���w�v�Z�@�ɔ�ׂČv�Z�ʂ�傫�����炷���Ƃ��ł��܂��B�܂��A����烂�m�}�[����у_�C�}�[�̌v�Z�͊��S�ɓƗ����Ă��邽�߁A���������������������܂��B
���ۂɉ�X��PAICS�ł��AMessage Passing Interface�iMPI�j�ɂ�郁�������U�^�̕����{����Ă���A���S�`����CPU��p��������v�Z���\�ɂȂ�܂��B
�}1�ɕ�������Ɋւ���x���`�}�[�N�̌��ʂ������܂��B�g�p����CPU�̐���8�{�i16��128�j�ɂȂ�ƌv�Z���x��7.18�{�ƂȂ��Ă���A��������������B������Ă��܂��B
�܂��A���̓t�@�C���̍쐬��v�Z���ʂ���͂��邽�߂̃��[�U�[�C���^�[�t�F�[�X�ł���uPaicsView�v���J������Ă��܂��i�}2�j�B
PAICS�ɂ͌��݁A����Hartree-Fock�iRHF�j�@�A��Moller-Plesset�ۓ��iMP2�j�@�AMP2�@��Resolution of the identity�iRI�j�ߎ���K�p����RI-MP2�@[2,3]�A�Ǎ݉��O���𗘗p����LMP2�@[4]�A����ɎO����Moller-Plesset�ۓ��iMP3�j�@�����MP2��RI�ߎ���K�p����RI-MP3�@[5]����������Ă��܂��B �����̌v�Z���_��p���āA�G�l���M�[�E�G�l���M�[���z�ɉ����āA�d�q���x�E�Ód�|�e���V�����E�d��[6]�Ƃ����������ʂ��A��̋ߎ���FMO�@�Ɋ�Â��Čv�Z���邱�Ƃ��ł��܂��i�\1�j�B �}3�Ɏ������̂́ARI�ߎ���p�����ꍇ�Ɨp���Ȃ��ꍇ��MP2�G�l���M�[���z�̌v�Z���Ԃ̔�r�ł��B RI�ߎ������邱�ƂŌv�Z���Ԃ�啝�ɒZ�k�ł��邱�Ƃ��ł��܂��B ���̂悤�Ɍv�Z���Ԃ�Z�k���闝�_��A���S���Y���̊J�����A�v�Z���w�ɂ�����d�v�Ȍ����e�[�}�̈�ɂȂ�܂��B �܂��APAICS�Ɏ�������Ă��邱���̌v�Z���_�𗘗p���āA�Ǝ��̉�͕��@�̊J�����i�߂Ă��܂��B �Ⴆ�AFMO�@��LMP2�@��g�ݍ��킹�����q�ԑ��ݍ�p��͖@�ł���Fragment Interaction Analysis Based on Local MP2�iFILM�j[4][7]�́A���̕��q����CH-�Α��ݍ�p���-�Α��ݍ�p�Ƃ��������U�͂ɋN�����鑊�ݍ�p���ڍׂɉ�͂��邱�Ƃ��ł�����@�ł��B �܂��AFMO�@�Ɋ�Â��Čv�Z����镔���d�q���x�ƕ����Ód�|�e���V�����𗘗p����Visualization of the interfacial electrostatic complementarity�iVIINEC�j[8,9,10,11]���A��X�̌������œƎ��ɊJ�����Ă���^���p�N���Ԃ̑��ݍ�p����͂��邽�߂̕��@�ł��B |
|
{kind=link}
{kind=link}
{kind=link}
{kind=link}
|
�֘A�����F
[1] T. Ishikawa, Chem. Phys. Lett., 761 (2020) 138103. [2] H. Ozono and T. Ishikawa, J. Chem. Theory Comput. 17 (2021) 5600 [3] T. Ishikawa, et al, J. Phys. Chem. Lett., 12 (2021) 11267 [4] H. Ozono, K. Mimoto, T. Ishikawa, J. Phys. Chem. B, 126 (2022) 8415 [5] S. Kousaka, T. Ishikawa, J. Chem. Theory Comput. 20 (2024) 5164 �֘A�O�����g�F �� 2022�N�x JST��w���V�Y�Ƒn�o�v���O�����iSTART�j��w�E�G�R�V�X�e�����i�^�uPARKS�N�Ɗ����x���v���O�����v�o�C�I���i�J���̌��������������鐶�̕��q�ʎq���w�v�Z�v���O�����uPAICS�v�A��\�A2022�N�x�`2022�N�x �� �ߘa5�N�x AMED ���n�������v���O���� �ٕ���Z���^�����V�[�Y�i�V�[�YH�j�R�̈��i�J������������ʎq���w�v�Z�Ɋ�Â���������C���V���R�n��Z�p�̊J���A��\�A2023�N�x�`2023�N�x �� �ߘa5�N�x ��ʍ��c�@�l�ӂ������t�B�i���V�����O���[�v��ƈ琬���c�����J�������� �R�̈��i�̊J���R�X�g���y������C���V���R�Z�p�̎Љ�����A��\�A2023�N�x�`2024�N�x �� �ߘa6�N�x AMED ���n�������v���O���� �ٕ���Z���^�����V�[�Y�i�V�[�YH�jVHH��p�����R�̈��i�J�������������鎟����C���V���R�n��Z�p�̊m���Ǝ��p���A��\�A2024�N�x�`2024�N�x �֘A�����F �� ���q�ԑ��ݍ�p��͑��u�A���q�ԑ��ݍ�p��͖@�y�уv���O����, �ΐ�x�u, �剀�h�M, ����2022-139395 �� ���q�ԑ��ݍ�p��͑��u�A���q�ԑ��ݍ�p��͖@�y�уv���O����, �ΐ�x�u, ����2023-25282�i2023�N2��21���j �� ���q�ԑ��ݍ�p��͑��u�A���q�ԑ��ݍ�p��͕��@, �ΐ�x�u, PCT/JP2023/030775�i2023�N8��25���j �� ���q�ԑ��ݍ�p��͑��u�A���q�ԑ��ݍ�p��͕��@�y�уv���O����, �ΐ�x�u, PCT/JP2024/005667�i2024�N2��19���j |
{kind=link}
{kind=link}
|
�Q�l�����F
[1] K. Kuwata, et al, Proc. Natl. Acad. Sci. USA, 104 (2007) 11921 [2] K. Yamaguchi, et al, Nat. Biomed. Eng., 3 (2019) 206 [3] T. Ishikawa, et al, J. Comput. Chem., 30 (2009) 2594 [4] D. Ishibashi, et al, EBioMedicine, 9 (2016) 238 �֘A�O�����g�F �� 2020�N�x �Ȋw����������� ��Ռ����iB�j�i��ʁj�R�v���I�����ʂݏo���t�@�[�}�R�t�H�A���f���̍\�z�ƐV�K���Ö�̊J���i��\�F�ΐ�x�u�j2020�N�x�`2022�N�x �� 2023�N�x �Ȋw����������� ��Ռ����iB�j�i��ʁj���w�V���y�����ɗL���ȃt�@�[�}�R�t�H�A���f���̍\�z�@�̊J���ƍR�v���I����ւ̉��p�i��\�F�ΐ�x�u�j2023�N�x�`2025�N�x �� 2026�N�x �Ȋw����������� ��Ռ����iB�j�i��ʁj�v���I���a���Ö�̍����W�J���������鍂�����q���͊w�A�v���[�`�̊m���i��\�F�ΐ�x�u�j2026�N�x�`2028�N�x �֘A�����F �� �v���I���a���Ö�, �����, ���c���s, ���c���u, �ΐ�x�u, ����2018-177224�i2018�N9��21���j �� �v���I���a�\�h�E���Í�, �����, ���c���s, ���_�x��, �_�c��, �ΐ�x�u, ����2016-170349�i2016�N8��31���j, ���J2018-35099�i2018�N3��8���j |
{kind=link}
| ��ŏq�ׂ��悤�ɁA�����ǂ̑n��͔��ɏd�v�Ȍ����e�[�}�ł����A���������V�������������J�����Ă��A�����̏ꍇ�͑ϐ����l�������a�����������o�����Ă��܂��܂��B ���ɃC���t���G���U�Ȃǂ�RNA�E�C���X�́A�ψق̃X�s�[�h�������A������͂����Ɏg���Ȃ��Ȃ��Ă��܂����ꂪ����܂��B �����ʼn�X�́A�R���s���[�^���g���ē���̖�ɑ���ϐ��ψق�\�����錤�����i�߂Ă��܂��B |
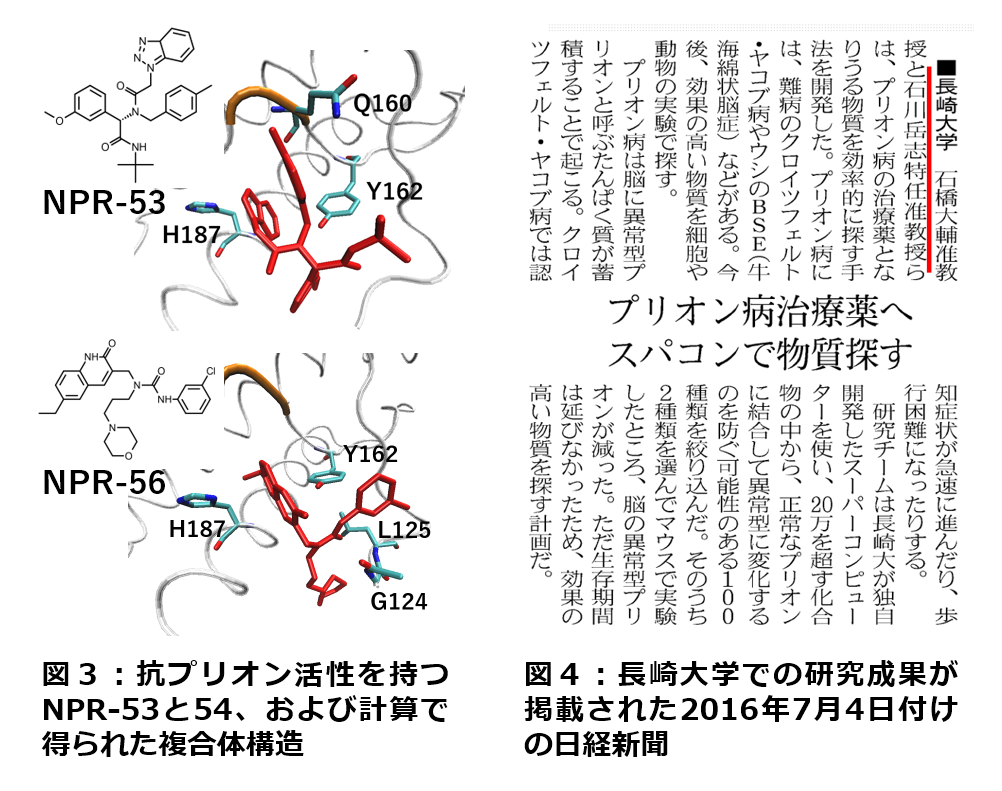
| ��X�́A�V�^�R���i�E�C���X�iSARS-CoV-2�j�̈��i�J���ɂ����g��ł��܂�[6]�B�h�b�L���O�v�Z�A���q���͊w�v�Z�A�t���O�����g���q�O���v�Z�Ƃ����������̃C���V���R�Z�p��p���āABP-1-102�Ƃ�����������SARS-CoV-2�̃��C���v���e�A�[�[�ƌĂ��^���p�N���̋@�\��j�Q���邱�Ƃ����o���܂����B���̌����́A��������w�̍H�w���E���w���E��[�Ȋw�������i�Z���^�[�E�q�g���g���E�C���X�w���������Z���^�[�Ƃ̋��������Ƃ��čs��ꂽ���̂ł��B |
|
�֘A�����F
[1] K. Watanabe, et al, Sci. Rep., 7 (2017) 9500 [2] J. N. Makau, et al, PLoS ONE, 12 (2017) e0173582 [3] J. N. Makau, et al, Viruses, 12 (2020) 337 [4] T. Ishikawa, et al, J. Phys. Chem. B, 122 (2018) 7970 [5] F. Mi-ichi, et al, PLOS Neglected Tropical Diseases, 13 (2019) e0007633 [6] T. Ishikawa, et al, J. Phys. Chem. B 129 (2025) 1740 �֘A�O�����g�F �� ����29�N�x ���{��Ì����J���@�\ �����nj����v�V�C�j�V�A�e�B�u�iJ-PRIDE�j �ԗ��A���[�o�g�ܗ�������Ӂh��W�I�Ƃ���j�Q�ܒT���\�S�e�𖾂Ǝ��Ö�J���ɂނ��ā\�i��\�F���s�����j2017�N�x�`2019�N�x �� ����29�N�x ���{��Ì����J���@�\ �����nj����v�V�C�j�V�A�e�B�u�iJ-PRIDE�j ��ܑϐ�RNA�E�C���X�o���\���@�̊m���Ɛv������̂��߂̃C���V���R�n��i��\�F���c���s�j2017�N�x�`2019�N�x �֘A�����F �� �R�E�C���X��, ���c���u, �����, �c���`��, ���R��, �ΐ�x�u, �r�c����, ���c��, ����2019-069458�i2019�N3��29���j �� PCT/JP2018/013592�i2018�N3��30���j�L�m���m������������эRRNA�E�C���X�� �� ����2017-72230 �i2017�N3��31���j�L�m���m������������эRRNA�E�C���X���Ö� |
|
��I���S���͕����̃O���R�[�X����Ɍ��������I���S���ł��B D-�O���R�[�X����-1,4�O���R�V�h�����Ŋ�\�����`�������V�N���f�L�X�g����(CD)�́A������100�N���O�ɔ�������A�Y�Ɖ��p���i��ł��܂��B ����AD-�O���R�[�X����-1,6�O���R�V�h�����Ŋ�\�����`�������V�N���f�L�X�g����(CI)�́A��30�N�O�ɔ�������܂������A�Y�Ɖ��p�͂��܂�i��ł��܂���B ��X�̌������ł́ACI�̎Y�Ɨ��p���߂����A���V����������Зl�ƌv�Z���w��p��������������i�߂Ă��܂��B | |
|
CI�̑�\�I�ȋ@�\�̈�����q��ڋ@�\�ł��B
CI�����ɗl�X�ȕ��q���ڂ��邱�ƂŁA�Q�X�g���q�Ɉ��萫��n���Ƃ������V���ȋ@�\��t�^���邱�Ƃ��ł���ƍl�����Ă��܂��B
�������ACD�ɔ�ׂČ����Ⴊ�ɒ[�ɏ��Ȃ��ACI�̕�ڃ��J�j�Y���̏ڍׂ͂قƂ�lj𖾂���Ă��܂���B
������CI�̕��q��ڋ@�\���𖾂��邽�߂ɁA���x�Ċ��@�A�v�Z���q���͊w�@�A�t���O�����g���q�O���@��p�����v�Z���s���܂���[1]�B
�ȉ��̓Q�X�g���q�Ƃ��ăR�G���U�C��Q10��I�������ꍇ�́ACI�i���j��CD�i�E�j�̕��ڏ�Ԃ̕��q���͊w�v�Z�̌��ʂł��B
�����̌��ʂ��ڍׂɉ�͂��邱�ƂŁACI��CD�̕��q���ڂ̃��J�j�Y���̈Ⴂ�𗝉����邱�Ƃ��ł��܂��i�ڍׂ͘_�����Q�Ƃ��ĉ������j�B
|
|
|
CI�̂�����̋@�\�́A�����ۂ̓��]�ڍy�f(GTF)��j�Q���邱�Ƃł��B
GTF�̓X�N���[�X���t���N�g�[�X�ƃO���R�[�X�ɕ������A�������ꂽ�O���R�[�X���d�����邱�ƂŁA�����̌����ƂȂ鑽���O���J���̍����𑣐i���܂��B
CI�́A���̃O���J��������j�Q���邱�Ƃ��m���Ă��܂��B����ACD�ɂ͂��̂悤�ȑj�Q�@�\�͂���܂���B�]���āA�O���J�������j�Q��CI�ɓ��L�̋@�\�Ƃ����܂��B
��X��CI�ɂ��GTF�̑j�Q���J�j�Y�����𖾂��邽�߂ɁA���q���͊w�@�A�t���O�����g���q�O���@�A���x�Ċ��@��p����������i�߂Ă��܂�[2]�B
�ȉ���GTF�̃O���J�������h���C���ƁACI�i���j�ECD�i�^�j�E�������i�E�j�̌�����Ԃ̕��q���͊w�v�Z�̌��ʂł��B
�����̌��ʂ��ڍׂɉ�͂��邱�ƂŁACI��GTF�j�Q���J�j�Y�����𖾂��邱�Ƃ��ł��܂��i�ڍׂ͘_�����Q�Ƃ��ĉ������j�B
|
|
�Q�l�����F
[1] W. Imamura, et al., Carbohydrate Polymers, 301A (2023) 120315 [2] W. Imamura, et al., Carbohydrate Polymer TA, 7 (2024) 100473 ���������F �� ���������i���V����������Ёj2020�N06��1���`2021�N11��30�� �� ���������i���V����������Ёj2022�N08��1���`2024�N09��31�� �� ���������i���V����������Ёj2024�N02��1���`2026�N03��31�� |
|
�����̐����E���ł��܂މ��w�����V�~�����[�V�����ł́A�o���I�ȃ|�e���V��������p����ÓT�v�Z�ł͂Ȃ��A�d�q�̉^���������ډ����ʎq���w�v�Z���K�v�ɂȂ�܂��B �������A�n�}���Ȃǂ̋ÏW�n�ł��̂悤�Ȍv�Z�����s���邽�߂ɂ́A�c��Ȍv�Z���Ԃ�v���邽�߁A��ʓI�ɂ͎��s������ł��B �����ʼn�X�́A�t���O�����g���q�O���@�𗘗p���Čv�Z���Ԃ�啝�Ɍ�������V�~�����[�V�����@���J�����܂���[1]�B �܂��A�ŏ��̉��p�����Ƃ��āA���n�t���̃��`���W�A�]�j�E���C�I���̉��������̃V�~�����[�V���������s���܂����B ���̍ہA150�ȏ�̐����q�Ɣ������q��S�ėʎq���w�I�Ɏ�舵���A�R���s���[�^�[�̒��ʼn��w�������Č����邱�Ƃɐ������܂����i�E�̓�����Q�Ɓj�B |
|
�Q�l�����F
[1] M. Sato, et al, J. Am. Chem. Soc. Comm. 130 (2008) 2396 |
{kind=link}
© 2018 ��������w ��w�@���H�w������ ���w��������w�H�w��U �����v�Z���w������